Welcome to the 3D-footprint tutorial. Here I will show how to make the most of the server. There are a few situations that you might find, please choose below the appropriate section to read more:
- you want to find human complexes in 3D-footprint (or any other keyword search)
- you just found a DNA motif in a promoter region and you want to know which protein might be binding to it
- there is a protein sequence that you believe is a DNA-binding protein and you want to find out what sort of motif it binds
- a difficult case
- an easy case
- you have the structure of a protein-DNA complex and you wish to calculate its structure-based PWM
1. How to find protein-DNA complexes associated to a keyword
A common way of querying 3D-footprint is by using keywords in the text form. This triggers a search for matches in the non-redundant 3D-footprint subset. The search covers a few slots of complexes in the database: the title, the reference, the title of redundant entries, SCOP/Pfam annotation and also the source (the scientific name of the organism of reference). For instance, a search with the term 'homeodomain' returns a few matches in title and SCOP annotation, such as:
matches in title:
| logo | matched complex |
|---|---|
| 1fjl_B: (...HOMEODOMAIN FROM THE DROSOPHIL...) |
matches in SCOP annotation:
| logo | matched complex |
|---|---|
| 1au7_A:lambda_repressor-like_DNA-binding_domains;Homeodomain-like; |
2. How to find proteins that recognize a similar DNA motif
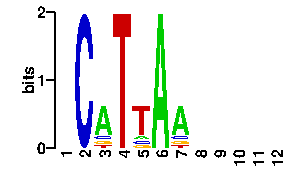
Say you are exploring promoter regions of a few genes that are of your interest, perhaps as they show similar expression patterns, and you suspect that a regulatory protein might be affecting the expression of all of them. Then you would probably search for some significant patterns or oligonucleotides, using your favourite software, and come up with a motif. If the motif is very conserved, then you can probably write it down as a string, such as TGTGA, but often motifs are degenerate and hence a more precise way of capturing them are Position Weight Matrices (PWMs). An example PWM for the TGTGA motif is now shown in PATSER/CONSENSUS format:
A | 0 0 0 0 7 C | 1 2 0 0 0 G | 0 6 0 8 0 T | 7 0 8 0 1
Any of these motif formats, including the TRANSFAC format, can be used to query 3d-footprint. Just paste the motif in the form and submit it. By default, motif searches perform local Smith-Waterman alignments, but you can also choose global alignments. Indeed, global alignments give better results when symmetrical or dimeric motifs are to be scanned. For instance, a local search will fail with the motif CGGNNNNNNNNNNNCGG (yeast Gal4) and the default E-value cutoff; however, a global search succesfully identifies complex 1d66_AB with an expectation value, reported by STAMP, in the order of 4e-09. What does this mean? It means that the protein binding motif CGGN{11}CGG is probably Gal4 if your promoters happen to be yeast promoters or, most likely, that the protein your are after is a Zn2/Cys6 DNA-binding domain with a very similar or identical binding interface. Finally, what can be said about this binding interface? Let us look at the interface figure. First, it is easy to see that most base pairs that make up the interface are color-filled, that is, they are labelled as indirectly readout bases. This means that they are partially recognized by means of sequence-specific DNA deformations. In addition, there are only three specific atomic interactions found at this interface (as seen by HBPLUS with default parameters), one in the first half and two in the second submotif. Readout PWMs are usually poor when so few contacts are found (see benchmark), but cumulative contact PWMs can still capture the right consensus, as this example illustrates.
3. How to find similar DNA-binding proteins (that bind known motifs)
3.1. A difficult case
Say you are working with E.coli FNR transcription factor, with this sequence:>P0A9E5|FNR_ECOLI Fumarate and nitrate reduction regulatory protein MIPEKRIIRRIQSGGCAIHCQDCSISQLCIPFTLNEHELDQLDNIIERKKPIQKGQTLFKAGDELKSLYAIRSGTIKSYTITEQG DEQITGFHLAGDLVGFDAIGSGHHPSFAQALETSMVCEIPFETLDDLSGKMPNLRQQMMRLMSGEIKGDQDMILLLSKKNAEERL AAFIYNLSRRFAQRGFSPREFRLTMTRGDIGNYLGLTVETISRLLGRFQKSGMLAVKGKYITIENNDALAQLAGHTRNVAThe first result produced by the server is a prediction of the putative interface residues along the (query) FNR sequence, which is obtained by piling up the BLAST alignment shown below. The more frequently a residue is aligned to interface residues in different non-redundant templates, the more confidence is given to the prediction:
| predicted signature | sequence context | ||
|---|---|---|---|
| RGTVETSRRKK | ...sprefrltmtRGdignylglTVETiSRllgRfqKsgmlavKgkyitiennd... |
| logo | E-value | signature | %Isim | Icover | organism | complex |
|---|---|---|---|---|---|---|
| - | 7e-80 | HHVTSY | 17.5 | 5/6 | DESULFITOBACTERIUM HAFNIENSE | 3e6c_C:CPRK OCPA DNA COMPLEX |
| - | 2e-71 | SRETNK | 36.9 | 6/6 | MYCOBACTERIUM TUBERCULOSIS | 3mzh_A:CRYSTAL STRUCTURE OF CAMP RECEPTOR ... |
| - | 2e-69 | QSRE | 6.2 | 4/4 | ESCHERICHIA COLI | 1zrf_A:4 CRYSTAL STRUCTURES OF CAP-DNA WIT... |
| - | 1e-12 | RTRTREH | -4.6 | 7/7 | ESCHERICHIA COLI | 1h9t_B:FADR, FATTY ACID RESPONSIVE TRANSCR... |
| - | 0.084 | LRSTQR | 15.0 | 6/6 | PSEUDOMONAS PUTIDA | 2xro_E:CRYSTAL STRUCTURE OF TTGV IN COMPLE... |
| 0.16 | SPTSQR | 55.1 | 6/6 | MYCOBACTERIUM TUBERCULOSIS | 1u8r_B:CRYSTAL STRUCTURE OF AN IDER-DNA CO... | |
| 0.23 | SPTSQRR | 55.1 | 6/7 | MYCOBACTERIUM TUBERCULOSIS | 2isz_B:CRYSTAL STRUCTURE OF A TWO-DOMAIN I... | |
| - | 0.30 | LAESTH | 4.5 | 5/6 | PYROCOCCUS HORIKOSHII | 2e1c_A:STRUCTURE OF PUTATIVE HTH-TYPE TRAN... |
| - | 0.36 | LYSYQSRHALA | 18.2 | 8/11 | ESCHERICHIA COLI | 1efa_B:CRYSTAL STRUCTURE OF THE LAC REPRES... |
| - | 0.57 | VR | -47.5 | 2/2 | HOMO SAPIENS | 1c9b_M:CRYSTAL STRUCTURE OF A HUMAN TBP CO... |
The first three hits show very small expectation values and contain "Winged helix" DNA-binding domains. The next hit is already a different SCOP superfamily, as you can see if you mouse over the hit name. The interface signature of all hits is coloured so that the user can check whether proteins with similar interfaces show similar DNA motifs. The second numeric column shows the percentage of interface similarity (%Isim), computed over the set of interface residues (those involved in atomic interactions or with heavy atoms <4.5Å away from nitrogen bases), highlighted in red in the alignments . This is an important number, as DNA-binding proteins with different interfaces will bind to different DNA motifs. Indeed 3D-footprint will only display the sequence logos of those complexes with at least 50 %Isim. The third numeric column further describes interface similarity, showing the fraction of the matched interface that is included in the BLAST sequence alignment, and it is useful to detect partial matches:
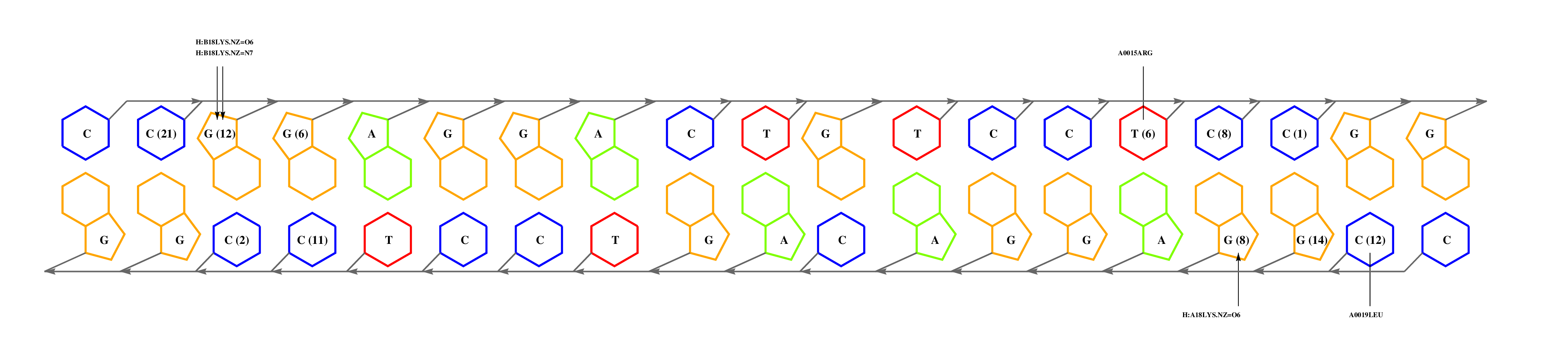
The question now is: are these matches equally significant? In this example we can choose to consider only alignments that cover complete interfaces, those with coverage values of 100%. Despite the smallest expectation value of 3e6c_C, the alignment missed one interface residue, which would still leave us with two matches. Before we continue, it can be noticed that 1zrf_A is actually a point-mutant, so perhaps we could use a redundant wild type complex, such as 1cgp_AB. If we inspect this interface, we can see that:
- GLU181 (glutamic acid in position 181) and 185ARG are responsible for recognition of a GA step, and they are conserved in FNR.
- ARG180, that hydrogen-bonds a G, is mutated for a valine, suggesting that the submotif preceding GA is different. In general, the effect of mutations on binding specificity can be further analyzed by inspecting the atomic interaction tables available in the download area, which provide a statistical description of the binding preferences of aminoacids.
--------HTHTVATA--ST--------------------YA L +---3e6c_C +----------------11 ----QT--STRGEC------RG-------------------- L ! +---1zrf_A
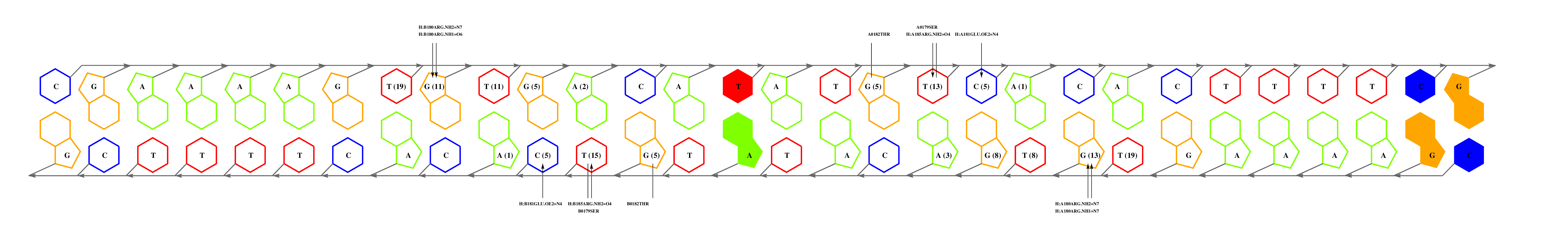
The second interesting match is 1u8r_B, which is aligned by BLAST with a relatively high expectation value but with a much higher interface similarity (55%). Inspection of this interface, together with the closely related next match (2isz_B), shows us that the residue with most interface contacts (GLN43) is not conserved. However, the conservation of 40THR40 suggests that the 5' end of the motif will be conserved. Our guess is that FNR binds a motif similar but not identical to TTAGGG.
What else can you do? It is always a good idea to check the literature, and section on
'motifs reported in the literature' points to some papers which show that indeed FNR binds a TTGAT motif,
which is indeed a chimera of the motifs of the best matches discussed above. In addition, as none of the matched complexes are sufficiently similar,
you might be interested in modelling the FNR binding interface using the
1cgp_AB complex as a template,
as done in this paper,
and you can do exactly that by following the link provided:
templates available => send to TFmodeller
After inspecting and evaluating the resulting model, you could then analyze the model's interface and derive structure-based PWMs as explained in the next section.
3.2. An easy case
Let us now suppose that you are working with this protein:>sp|P35891|DNAA_SALTY initiator protein dnaA Salmonella typhimurium MSLSLWQQCLARLQDELPATEFSMWIRPLQAELSDNTLALYAPNRFVLDWVRDKYLNNIN GLLNTFCGADAPQLRFEVGTKPVTQTLKTPVHNVVAPAQTTTAQPQRVAPAARSGWDNVP APAEPTYRSNVNVKHTFDNFVEGKSNQLARAAARQVADNPGGAYNPLFLYGGTGLGKTHL LHAVGNGIMARKPNAKVVYMHSERFVQDMVKALQNNAIEEFKRYYRSVDALLIDDIQFFA NKERSQEEFFHTFNALLEGNQQIILTSDRYPKEINGVEDRLKSRFGWGLTVAIEPPELET RVAILMKKADENDIRLPGEVAFFIAKRLRSNVRELEGALNRVIANANFTGRAITIDFVRE ALRDLLALQEKLVTIDNIQKTVAEYYKIKIADLLSKRRSRSVARPRQMAMALAKELTNHS LPEIGDAFGGRDHTTVLHACRKIEQLREESHDIKEDFSNLIRTLSSIf you use the protein form and submit the DNAA sequence, the server returns you a list of similar complexes in 3D-footprint:
| logo | E-value | signature | %Isim | Icover | organism | complex |
|---|---|---|---|---|---|---|
| 1e-48 | RLPDHTTLH | 100.0 | 9/9 | ESCHERICHIA COLI | 1j1v_A:CRYSTAL STRUCTURE OF DNAA DOMAINIV ... | |
| 2e-40 | KPDHTMY | 72.7 | 7/7 | MYCOBACTERIUM TUBERCULOSIS | 3pvv_B:STRUCTURE OF MYCOBACTERIUM TUBERCUL... |
The first hit corresponds to E.coli DnaA, a very close relative of Salmonella, and it has an identical interface and an almost identical protein sequence. Hence, we could safely use the structure-based PWM of 1j1v_A to scan Salmonella genomic sequences if we need to. For that purpose please click on the scan! link and a form will appear in which you can apply the set of RSA-Tools in order to look for matches of the DnaA motif in DNA sequences. You can either scan particular genomic regions of interest or the available set of complete genomes (with PATSER). Please read this protocol for more details.
4. How to calculate a structure-based position weight matrix
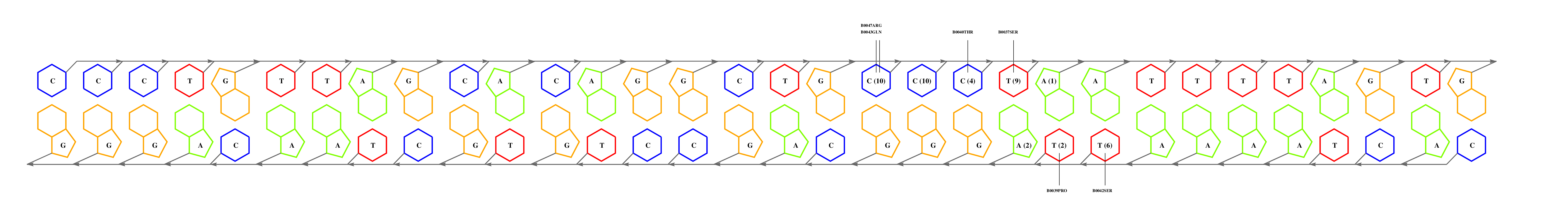
If you have the coordinates of a protein-DNA complex in PDB format, obtained either experimentally (ideally) or after modelling exercises, then you may want to use 3D-footprint to calculate a position weight matrix that captures binding specificity. All you need to do is prepare a PDB file, remove all DNA chains but two of them, and upload it at the interactive footprint form. What the server will do with your file is summarized in this flowchart, leaving out the PubMed exploration. The most relevant parameters that you can change in this form are rotamer sampling and relaxed hydrophobics. Interface side-chain rotamers can be sampled to minimize the chance of missing high scoring atomic interactions, although this is usually not necessary for crystallographic models. Water molecules need to be removed during rotamer sampling, therefore users are advised to turn it off when seeking for conserved water-mediated hydrogen bonds. Relaxing the distance cutoff for hydrophobic contacts is usually only recommended for the analysis of comparative models of protein-DNA complexes, since models often contain errors which could mask otherwise highly scoring interface interactions.
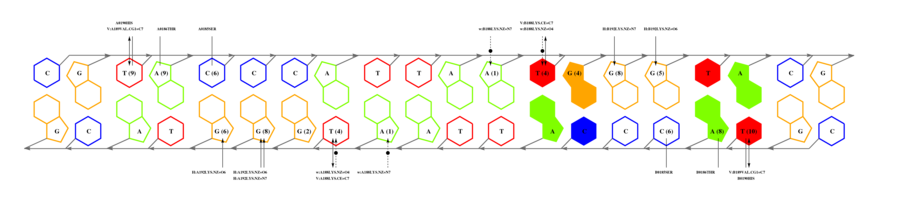
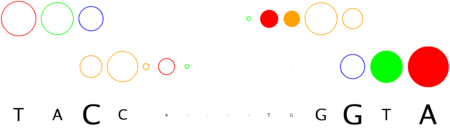
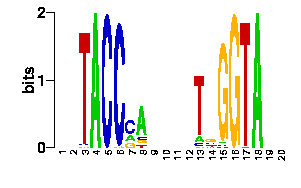
We will now submit a job with the sample PDB file provided here. Results will typically look like this (please pay attention to the specificity estimate, in a SCOP superfamily scale):
# finding interface residues/bases (chain:number:Bfactor) ... A: 186 : 23.31 B: 189 : 22.69 A: 190 : 23.71 <== usually interface side chains have small Bfactors A: 192 : 23.99 B: 192 : 23.67 [...] # scanning interface rotamers ... # model 0 contains 132 residues # Reading _dna_dd40b0-interface_scwrl_amf.pdb... # strand1 (D) contains 20 bases # strand2 (C) contains 20 bases # DNA sequence complementary region (20, ?=too distant base pairs): CGTACCCATTAATGGGTACG |||||||||||||||||||| GCATGGGTAATTACCCATGC # Cannot parse interface HET atoms file _het_dd40b0-interface_scwrl_amf.pdb , skip it <== this appears when rotamer sampling is ON # Calculating protein-dna interface scores... # Original PDB DNA sequence... <== atomic interactions found in the original coordinate set # Original interface contacts: : H : LYS NZ A0192 <- 2.86 -> DG N7 C0015 : score 5.15407 : H : LYS NZ A0192 <- 2.91 -> DG O6 C0015 : score 5.41285 : H : LYS NZ A0192 <- 3.05 -> DG O6 C0016 : score 5.25788 : H : LYS NZ B0192 <- 3.22 -> DG N7 D0035 : score 4.77847 : H : LYS NZ B0192 <- 3.28 -> DG O6 D0036 : score 5.00329 : V : VAL CG1 B0189 <- 3.74 -> DT C7 C0003 : score 4.87616 A <== rotamer A gave highest score, might appear if rotamer sampling is ON : V : LYS CE A0188 <- 3.83 -> DT C7 C0013 : score 2.552 : V : VAL CG1 A0189 <- 3.63 -> DT C7 D0023 : score 5.00828 A : V : LYS CE B0188 <- 3.85 -> DT C7 D0033 : score 2.53914 seq orig PDB 0 cgTaCCcAttaaTgGGtAcg 40.5821 1.0 total 40.5821 step 15.19 bp 4.15 dGDNA 23.22 <== uppercase bases get contactsInterface of input file ?
(reduced version)legend
legendEstimated binding specificities ?
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| readout + contact |
|
{kind=link}
{kind=link}